Spongiforme Enzephalopathien (Prionkrankheiten) sind solche Krankheiten, bei denen pathologische Formen von Prionproteinen an der Entwicklung beteiligt sind. Wir wissen immer mehr über Prionkrankheiten, aber die Schlüsselaspekte sind unbekannt - derzeit verfügt die Medizin nicht über die Mittel, um Patienten von diesen Krankheiten zu heilen.
Spongiforme Enzephalopathien, d. H. Prionkrankheiten, können sich während des Lebens entwickeln, während andere aus vererbten Genmutationen entstehen, die von Geburt an vorhanden sind. Innerhalb dieser Gruppe treten beim Menschen mehrere Entitäten auf, beispielsweise die Creutzfeldt-Jakob-Krankheit oder tödliche familiäre Schlaflosigkeit.
Prionkrankheiten sind seit langem sehr mysteriös. Im Gegensatz zu anderen Krankheitserregern wie Bakterien, Viren oder Pilzen enthalten sie keine Nukleinsäure - Prionen bestehen nur aus Proteinen. Die Theorie der Prionkrankheiten wurde von S. Prusiner entdeckt, diese Entdeckung wurde in der wissenschaftlichen Gemeinschaft sehr geschätzt - 1997 erhielt der Forscher den Nobelpreis für Medizin. Obwohl seit der Geburt des Prion-Konzepts relativ viele Jahre vergangen sind, glauben einige Wissenschaftler immer noch, dass es unvollständig ist, und untersuchen die Natur dieser Zustände weiter - einige der Faktoren, die für spongiforme Enzephalopathien verantwortlich sind, wurden nun bestätigt.
$config[ads_text1] not found
Prionkrankheiten: Ursachen
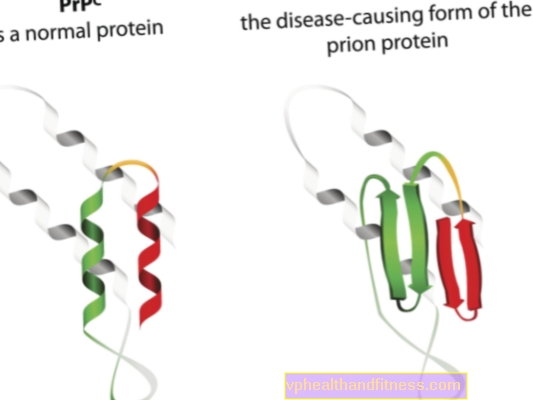
Die Ätiologie von Prionkrankheiten hängt mit der Umwandlung normaler Prionproteine in pathogene, pathogene Formen zusammen. Prionen sind Proteinmoleküle, die im Körper eines jeden Menschen vorkommen. Ihre Funktion ist noch nicht ganz klar, aber es ist bekannt, dass Prionproteine unter normalen Bedingungen den Körper nicht schädigen. Anders sieht es aus, wenn Prionen ihre Struktur ändern und zu pathogenen Partikeln werden - dann entwickelt sich eine von mehreren spongiformen Enzephalopathien. Im Körper natürlich vorkommende Prionen werden als PRPC bezeichnet, während abnormale Formen als PRPSC bezeichnet werden. Letztere sind nicht nur deshalb ein ernstes Problem, weil sie sich in Form von Ablagerungen im Nervengewebe ansammeln und dessen Schaden verursachen können, sondern auch, weil sie normale Prionen in eine missgebildete Form umwandeln können (einfach gesagt, PRPSC) kann normale Proteine mit ihrem pathogenen Potential "infizieren").
Lesen Sie auch: Huntington-Krankheit (Huntington-Chorea): Ursachen, Symptome, Behandlung Muskelzittern - Ursachen. Was bedeutet Muskelzittern? Krankheiten, die am schnellsten töten: SCHOCK, EBOLA, VERDAMMT, ANGRIFF, NOTFALL [GALE ...Grundsätzlich gibt es 3 Ursachen für spongiforme Enzephalopathien:
$config[ads_text2] not found- sporadisch (pathogene Mutation tritt in somatischen Zellen auf, sie tritt während des Lebens des Patienten auf),
- Familie (resultierend aus der Last der von den Eltern geerbten Mutationen),
- Passage (im Zusammenhang mit der Einführung pathogener Prionen in den menschlichen Körper, z. B. durch mit diesen Partikeln kontaminierte Wachstumshormonpräparate oder Hornhauttransplantation von einer Person, die an einer spongiformen Enzephalopathie leidet).
Spongiforme Enzephalopathien: Creutzfeldt-Jakob-Krankheit
Die Creutzfeldt-Jakob-Krankheit (CJD) wurde erstmals in den frühen 1920er Jahren beschrieben. Es gibt 4 Arten der Krankheit:
- sporadische CJD (die häufigste, die bis zu 9/10 aller CJD-Fälle ausmacht)
- Heimatstadt von CJD
- CJD mit Gürtel
- Variante von CJD
Das Krankheitsbild im Verlauf verschiedener Varianten der Creutzfeldt-Jakob-Krankheit kann variabel sein. Die häufigsten Erkrankungen im Verlauf dieser Gruppe spongiformer Enzephalopathien sind:
- Demenzstörungen (einschließlich fortschreitender Verschlechterung des Gedächtnisses, der Aufmerksamkeit und der Konzentration)
- Myoklonus (unwillkürliche Bewegungen wie plötzliche Muskelzuckungen)
- Kleinhirnfunktionsstörung (manifestiert sich beispielsweise durch Gleichgewichtsstörungen)
- verschwommene Sicht
- pyramidenförmige und extrapyramidale Symptome
Im Verlauf von CJD-Varianten können auch psychische Störungen (z. B. Angstzustände, depressive Verstimmungen), Schmerzen und andere unwillkürliche Bewegungen als die oben genannten auftreten.
$config[ads_text3] not found
Die Prognose für die Creutzfeldt-Jakob-Krankheit ist schlecht - beispielsweise dauert es bei Patienten mit sporadischer CJD durchschnittlich vier bis fünf Monate vom Auftreten der Krankheitssymptome bis zum Tod.
Spongiforme Enzephalopathien: Gerstmann-Straussler-Scheinker-Syndrom
Das Gerstmann-Straussler-Scheinker-Syndrom (GSS) tritt normalerweise in Familien auf und wird durch eine vererbte Mutation im PRNP-Gen verursacht. Es wird als die am langsamsten fortschreitende spongiforme Enzephalopathie angesehen. Das GSS-Team besteht aus:
- spinocerebelläre Ataxie
- Dysarthrie
- Demenzerkrankungen
- Schluckstörungen
- Nystagmus
- erhöhte Muskelspannung
Patienten, bei denen GSS diagnostiziert wurde, haben eine variable Zeitdauer, und bei einigen Patienten tritt der Tod mehr als 10 Jahre nach Beginn auf.
Spongiforme Enzephalopathien: tödliche familiäre Schlaflosigkeit
Tödliche familiäre Schlaflosigkeit ist eine Prionkrankheit, die durch Mutationen im PRNP-Gen verursacht wird. Die Krankheit ist äußerst selten und wurde bisher in 28 Familien weltweit diagnostiziert. Im Verlauf einer tödlichen familiären Schlaflosigkeit ist das erste Symptom die Unfähigkeit zu schlafen. Dieses Problem führt zu Angststörungen und Halluzinationen des Patienten. Die Folge des ständigen Mangels an Nachtruhe ist eine Funktionsstörung des autonomen Systems (einschließlich Veränderungen der Herzfunktion, Schwitzen und Störungen des Verdauungssystems). Außerdem nimmt das Körpergewicht progressiv ab. In fortgeschritteneren Stadien tödlicher familiärer Schlaflosigkeit treten hormonelle Störungen auf, und im Verlauf der Krankheit treten Symptome einer Demenz auf.
Die Prognose für tödliche familiäre Schlaflosigkeit ist wie für andere spongiforme Enzephalopathien schlecht: Patienten sterben normalerweise innerhalb von drei Jahren nach Beginn.
Spongiforme Enzephalopathien: Prionopathie mit variabler Anfälligkeit für Protease
Das Auftreten der diskutierten spongiformen Enzephalopathien hängt hauptsächlich mit Mutationen im PRNP-Gen zusammen. Diese Mutationen betreffen jedoch verschiedene Codons dieses Gens, und daher werden mehrere verschiedene Prionkrankheiten unterschieden. Eine relativ neu beschriebene Einheit (2008) ist die Prionopathie mit variabler Anfälligkeit für Protease. Menschen, die an dieser Krankheit leiden, tragen Mutationen in bis zu drei Codons des PRNP-Gens.
Bei Prionopathie mit variabler Proteaseempfindlichkeit erfahren die Patienten:
- kognitive Beeinträchtigung
- extreme Schwere psychiatrischer Störungen: Sie können Euphorie und Unruhe sein, aber auch signifikante Apathie
- Dysarthrie
- Aphasie (Störungen der Sprachfunktionen)
Die durchschnittliche Krankheitsdauer bei dieser Prionopathie beträgt weniger als 4 Jahre.
Spongiforme Enzephalopathien: kuru
Kuru gilt heute als eine Krankheit, die praktisch nicht mehr existiert - sie wurde bei Vertretern von Stämmen aus Papua-Neuguinea gefunden, die Kannibalismus praktizierten. Das dominierende Symptom dieser spongiformen Enzephalopathie ist die progressive Kleinhirnataxie. Es kann von unwillkürlichen Bewegungen (hauptsächlich in Form von Chorea, Zittern und Athetose) sowie von Harn- und Stuhlinkontinenz begleitet sein. Patienten, die an Kuru leiden, erfahren ebenfalls signifikante Stimmungsschwankungen, sie entwickeln primitive Reflexe (z. B. Saugen). Ein charakteristisches Problem bei dieser Prionkrankheit sind erzwungene Weinen oder Lachen - aufgrund der letzteren Phänomene wird Kuru manchmal als "lachender Tod" bezeichnet.
Spongiforme Enzephalopathien: Diagnose
Prionkrankheiten können aufgrund der Symptome des Patienten vermutet werden. Sie sind jedoch ziemlich unspezifisch, da sie auch im Verlauf einer Reihe anderer Krankheiten auftreten können, die nicht mit Prionen zusammenhängen. Aus diesem Grund wird bei der Diagnose von spongiformen Enzephalopathien auch Folgendes verwendet:
- Bildgebungstests (z. B. Magnetresonanztomographie, die es ermöglicht, Veränderungen im Zusammenhang mit der Degeneration des Gehirns durch Prionproteine zu erfassen),
- Labortests (wie die Beurteilung von Proteinkonzentrationen in der Cerebrospinalflüssigkeit, z. B. MAP-Tau-, S-100- oder 14-3-3-Proteine),
- Gentests (um das Vorhandensein von Mutationen beim Patienten festzustellen),
- immunhistochemische Tests (unter Verwendung von Antikörpern gegen Prionproteine).
Die Diagnose kann auch durch eine Autopsie des Gehirns bestätigt werden, bei der Veränderungen festgestellt werden können, die für spongiforme Enzephalopathien charakteristisch sind. Dies können schwammige Läsionen sein, die unterschiedlich verteilt sind und eine unterschiedliche Struktur (abhängig von einer bestimmten Krankheitseinheit), Amyloidplaques und neuronale Defekte aufweisen.
Spongiforme Enzephalopathien: Behandlung
Prionkrankheiten sind derzeit nicht heilbar - trotz zahlreicher Studien, die seit vielen Jahren durchgeführt werden, gibt es in der Medizin immer noch keine Medikamente, die ihren Fortschritt verlangsamen oder vollständig hemmen könnten. Die symptomatische Behandlung wird bei Patienten mit spongiformen Enzephalopathien angewendet, um die Intensität der Symptome zu lindern und ihre Lebensqualität so weit wie möglich zu verbessern.
Die Arbeiten zur Behandlung spongiformer Enzephalopathien dauern jedoch noch an. Wissenschaftler versuchen, verschiedene Methoden anzuwenden - das erste Beispiel ist die Gentherapie. Sie würden die Nukleinsäuren und die in ihrer Struktur vorhandenen Mutationen beeinflussen - der Zweck der Anwendung der Gentherapie wäre die Neutralisierung von Fehlern im genetischen Code. Ein anderer Ansatz ist die Grundlage der Immuntherapie. Derzeit wird daran gearbeitet, Antikörper zu entwickeln, deren Aufgabe es wäre, pathogene Prionen zu eliminieren. Eine andere Methode, die das Potenzial zur Bekämpfung spongiformer Enzephalopathien sieht, ist die Behandlung mit synthetisierten Proteinmolekülen, die, sobald sie in den Körper des Patienten eingeführt werden, pathologische Proteine neutralisieren würden.
Empfohlener Artikel:
Enzephalopathien - Ursachen, Arten und Symptome