Die Fabry-Krankheit ist eine seltene genetisch bedingte Krankheit, die Symptome wie Degeneration der Gelenke oder Herzerkrankungen aufweist. Dies macht es viel schwieriger, eine frühzeitige Diagnose zu stellen und eine geeignete Behandlung durchzuführen, die sogar zu einer Behinderung führen kann, nicht nur zu einer motorischen Behinderung. Was sind die Ursachen und Symptome der Fabry-Krankheit? Was ist sein Erbe? Ist es möglich, es zu heilen?
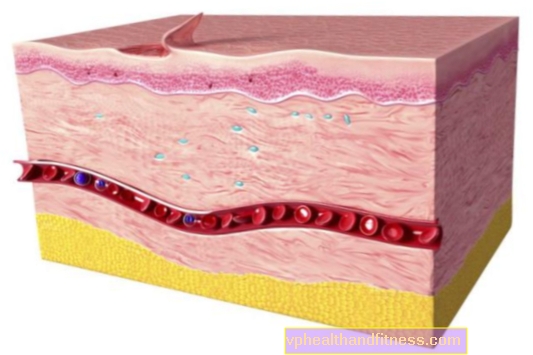
Die Fabry-Krankheit ist eine angeborene Speicherkrankheit aus der Mucopolysaccharidgruppe. Dies bedeutet, dass in seinem Verlauf Glycosphingolipide (schädliche Stoffwechselprodukte) in Blutgefäßen und anderen Geweben sowie in Organen abgelagert (gelagert) werden und somit - deren Funktionen beeinträchtigt werden. Dieser Prozess ist auf das Fehlen des Alpha-Galactosidase A-Enzyms zurückzuführen, das für die Entfernung von Glycosphingolipiden aus dem Körper wesentlich ist.
$config[ads_text1] not found
Die Häufigkeit der Krankheit wird auf 1: 40.000–1: 120.000 männliche Geburten geschätzt. Somit gehört die Krankheit zur Gruppe der seltenen Krankheiten. Die Fabry-Krankheit betrifft nach Schätzungen der Patientenvereinigung 50 bis 100 Menschen in Polen.
Fabry-Krankheit - Ursachen und Vererbung
Die Krankheit wird durch eine Mutation im GLA-Gen am Xq22-Locus verursacht, das das Protein des oben genannten Alpha-Galactosidase-Enzyms codiert. Das mutierte Gen befindet sich auf dem X-Chromosom, wird jedoch nicht wie die meisten Krankheiten in dieser Gruppe vererbt, d. H. Autosomal rezessiv (wie von den meisten Quellen berichtet), sondern als dominantes geschlechtsgebundenes Merkmal, ähnlich der Hunter-Krankheit und der Danon-Krankheit (Quelle: Kucharczyk-Foltyn) UND., Herzmanifestation der Fabry-Krankheit - der Weg zur Diagnose. Ein Fallbericht, "Folia Cardiologica Excerpta" 2011, Band 6, Nr. 2).
Fabry-Krankheit - Symptome
Symptome der Krankheit sind paroxysmale Schmerzen in den Gliedmaßen (insbesondere in den Gelenken), die den Verdacht auf eine rheumatische Erkrankung erwecken. Es gibt zwei Arten von Schmerzen im Verlauf der Krankheit:
Das häufigste Symptom der Fabry-Krankheit sind Störungen der Körperthermoregulation - Schwitzen. Dies bedeutet, dass sich der Körper nicht von selbst abkühlen kann, was zu einer schnellen Überhitzung führt und Fieber und Schmerzen in den für die Krankheit charakteristischen Füßen und Händen verursacht.
- Akroparästhesie - Schmerzen in Händen und Füßen, die meist in Form von Brennen und Juckreiz auftreten und tagsüber regelmäßig auftreten;
- die sogenannteFabry-Durchbrüche sind Anfälle von starken Schmerzen in den Füßen und Händen, die auf die übrigen Gliedmaßen ausstrahlen. Angriffe können von Minuten bis zu Tagen dauern.
Die beschriebenen Schmerzsymptome resultieren aus der Ablagerung von Glycosphingolipiden in den Zellen des Nervensystems.
$config[ads_text2] not foundThermoregulationsstörungen (Patienten schwitzen nicht) und ein charakteristischer Ausschlag in Form von keratotischen Hämangiomen treten im Bereich der Oberschenkel, der Leistengegend, der Genitalien und des Abdomens auf.
Die Anreicherung von Glycosphingolipiden, hauptsächlich in den Gefäßendothelzellen, schädigt viele Organe, insbesondere die Nieren, das Herz und das Gehirn, was sich manifestiert durch:
- Degeneration in der Hornhaut des Auges, die durch die Speicherung von Glycosphingolipiden in den Blutgefäßen verursacht wird;
- Komplikationen aus dem Kreislaufsystem: hypertrophe Kardiomyopathie, Herzinsuffizienz, Arrhythmie, Herzklappeninsuffizienz, Herzinfarkt,
- Nierenversagen und Proteinurie;
- Schwindel und Ohnmacht (das Schlaganfallrisiko steigt ebenfalls);
- Magenschmerzen durch Magen-Darm-Erkrankungen. Normalerweise sind dies postprandiale Schmerzen, Durchfall und Übelkeit
Obwohl die ersten Symptome der Krankheit bereits in der Kindheit auftreten, war dies bei 75% der Fälle der Fall Befragte - bis zu 73 Prozent Diagnosen werden nach mindestens 5 Jahren Suche gestellt.
Fabry-Krankheit - Behandlung
Die kausale Behandlung genetisch bedingter Krankheiten ist nicht möglich. Die Therapie basiert auf einer enzymatischen Ergänzung, d. H. Der Abgabe von Präparaten, die das fehlende Enzym enthalten, üblicherweise auf intravenösem Weg, um die bereits vorhandenen Speicherablagerungen zu entfernen und die Bildung neuer Ablagerungen zu verhindern. Zwei Präparate, die Alpha-Galactosidase A enthalten (Agalsidase a und Agalsidase b), sind in der Europäischen Union registriert und beide in Polen erhältlich.
$config[ads_text3] not foundWeitere Informationen zur Krankheit finden Sie auf der Website der Vereinigung der Familien mit Fabry-Krankheit.
Wichtig
Die Fabry-Behandlung wird in Polen nicht erstattet
In Polen kämpfen Patienten seit über 12 Jahren erfolglos um den Zugang zu einer erstatteten Therapie, was das Fortschreiten der Krankheit verlangsamt und lindert und den Patienten die Chance auf ein relativ normales und aktives Leben gibt. Derzeit haben sich nur Personen für das sogenannte qualifiziert Wohltätigkeitsprogramme von Arzneimittelherstellern. Diese Form des Zugangs zur Behandlung wird von Personen genutzt, die an klinischen Studien teilgenommen haben, sowie von Patienten in schwerem Zustand. Sie sind sich jedoch nicht sicher, wie lange die gemeinnützige Finanzierung der Therapie dauern wird. Viele neu diagnostizierte Patienten - einschließlich Kinder, deren Krankheit sich noch nicht nachhaltig verändert hat - haben keine Chance auf eine lebensrettende Behandlung. Unter den an der Studie teilnehmenden Personen erhalten 21 Patienten eine wohltätige Behandlung.
- Trotz der Existenz einer Enzymersatztherapie, die die Entwicklung der Krankheit stoppt und lebensbedrohliche Komplikationen vermeidet, bleiben viele polnische Patienten ohne Behandlung. In allen Ländern der Europäischen Union erhalten Menschen mit Morbus Fabry eine wirksame und erstattete Behandlung. Es scheint, dass nur in Polen unser Recht auf Leben für das Gesundheitssystem zu teuer ist, um es zu berücksichtigen, erklärt Roman Michalik vom Verband der Familien mit Fabry-Krankheit.
Lesen Sie auch: HUNTER-SYNDROM - Symptome und Behandlung von Mukopolysaccharidose Typ II VIBRANTES CHROMOSOM X-Syndrom (FRA X) - Symptome und Diagnose des FRA X-Syndroms Mukopolysaccharidose: Ursachen, Symptome und Behandlung